Observation-by-feature support has arrived
Since the start of the Vitessce project in late 2018, our team has been focused on addressing the challenges of interactive web-based visualization of multi-modal and spatial single-cell datasets.
However, this problem has a broad scope and we've had to prioritize certain features over others: we have 142 open feature requests. This broad scope, coupled with the initial set of technologies used to generate data in the first phase of the HuBMAP Consortium (the first user of Vitessce), meant that we initially developed many features for scRNA-seq and bioimaging datasets. Early on, we also discovered a gap in web-based tools for dynamic rendering of bioimaging data on the client side, so members of our team took a detour to develop the Viv library. Meanwhile, the HuBMAP Consortium moved into its second phase, adding new labs and contributors, and generating a wide range of multi-modal datasets using technologies like SNARE-seq.
In parallel, the single-cell community is generalizing the cell-by-gene matrix from transcriptomics to represent other types of experimental data using "observation-by-feature" matrices (abbreviated FOM in the Feature and Observation Matrix Schema Working Group terminology). This is enabling an ecosystem of tools to be developed around the data structure, such as AnnData and Scanpy. Emerging formats such as MuData extend this paradigm yet further to support multi-modal use cases via container data structures that hold multiple observation-by-feature matrices. Multiple observation-by-feature matrices may share the feature axis (multi-batch), the observation axis (multi-modal), neither axis (diagonal), or some combination (mosaic). Our tools should support visualization of datasets that conform to any of these four cases.
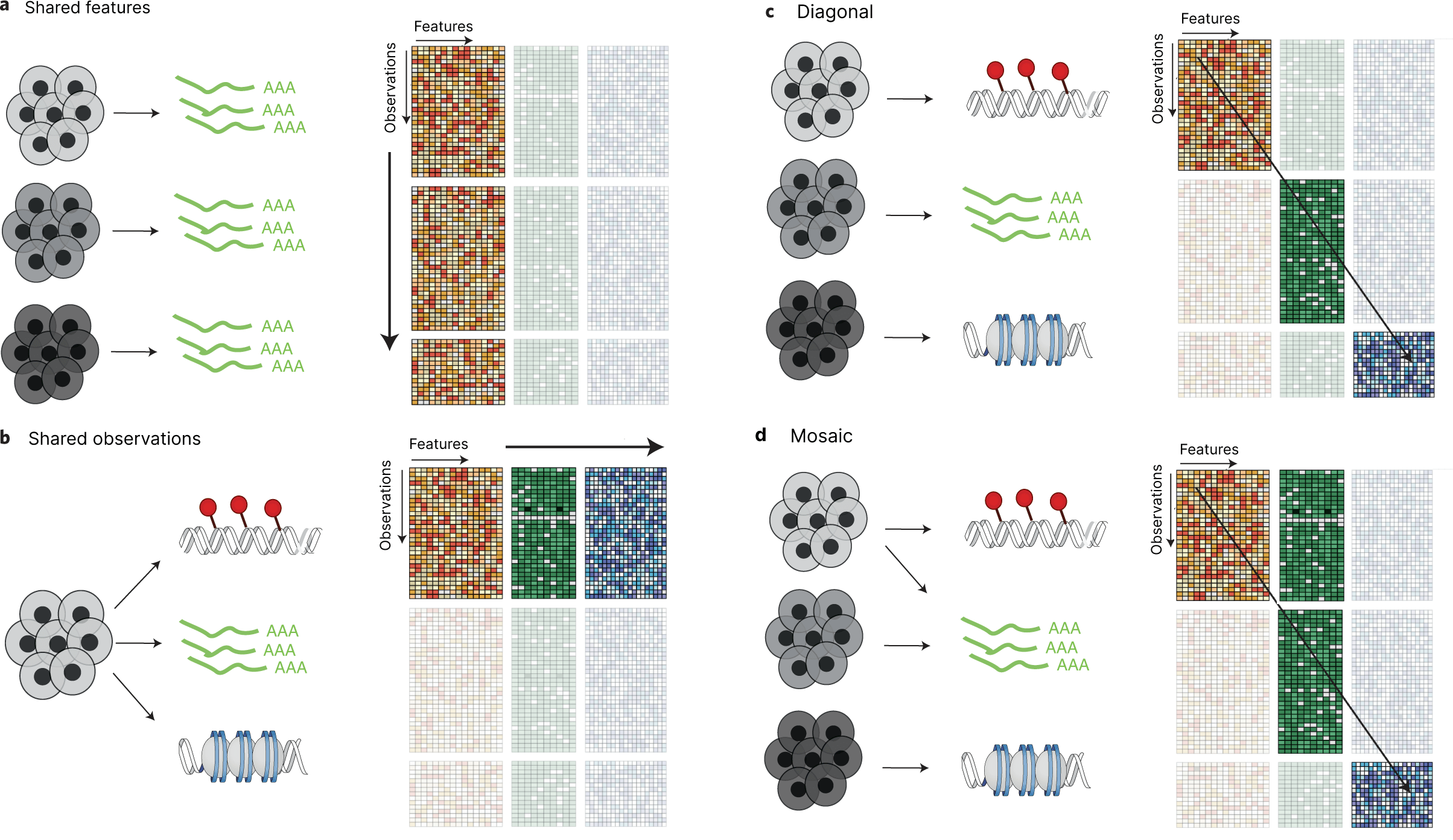
Adapted from Figure 1, Argelaguet et al., Nature Biotechnology 2021
Updates in Vitessce
Vitessce was initally coupled to the more specific cell-by-gene ideas, preventing straightforward use with non-transcriptomics or multi-modal datasets. We needed a major update to realize the FOM paradigm in Vitessce. In our latest update, we have changed how we represent data and how we configure mappings between data and views in order to support the visualization of datasets containing multiple observation-by-feature matrices.
Data types
Data types no longer assume observation types like cell.
Instead, to facilitate re-use across observation types, we have split up each of the former data types into multiple more minimal data types.
For example, obsLocations stores (x, y) coordinates per observation, obsEmbedding stores (dim1, dim2) coordinates per observation for a given embedding type, and obsSegmentations stores polygon vertices per observation.
Simplified data types should also make it easier to implement new file types (including via plugins).
The former raster data type has been split into two new data types, image and obsSegmentations, to better reflect semantics rather than exposing the implementation differences between bitmask and polygon segmentations. (Internally, the fact that an obsSegmentations file type may return either polygons or bitmasks is handled by returning an obsSegmentationsType from the data loader class.)
For the full list of data types, please visit the updated Data Types and File Types page.
Dataset definitions
Each of the datasets in the view configuration may now contain multiple file definitions corresponding to the same data type.
Previously, dataset definitions were limited to one file per data type.
New coordination types
The new coordination types obsType, featureType, and featureValueType enable "entity types" to be specified.
For example, if we have a cell-by-peak matrix from an scATAC-seq experiment, then the entities along the observation axis are cells and the entities along the feature axis are peaks:
dataset: 'my_atac_experiment',
obsType: 'cell',
featureType: 'peak',
featureValueType: 'count'
With these coordination types:
- more accurate strings like
100 cells x 200 peakscan be rendered in the interface (as opposed to the more abstract100 observations x 200 features). - specific values like
featureType: 'gene'can be used to offer specific analysis functionality (e.g., differential expression tests that are valid only for transcriptomics experiments). - the mapping between files and views can leverage both data types and entity types, as described in the next section.
Mapping between files and views
File lookup from within a view now takes into account
(dataset, dataType, viewCoordinationValues).
The viewCoordinationValues refers to the coordination values that a particular view takes on at a given point in time.
This is required to disambiguate between multiple files within a dataset which have the same dataType (now that each dataset may contain multiple files per data type).
The diagram below shows how the mapping between views and files would be performed for a configuration containing one dataset and three views.
- Heatmap
- Scatterplot
- Spatial
- Hide matches




Joint file types
Joint file types can now be defined via functions which expand one file definition into an array of file definitions.
Name changes
View type names have been updated to use obs and feature terminology (e.g., cellSets is now obsSets and genes is now featureList).
Coordination type names have been updated to use obs and feature terminology (e.g., cellColorEncoding is now obsColorEncoding and geneExpressionColormap is now featureValueColormap).
Exported constants
Exported constants have been updated to reflect the changes to view types, file types, data types, and coordination types as described here.
File definitions
File definitions no longer require the type (data type) property.
Instead, we now map file types to data types internally.
View config schema
The changes described in this blog post are reflected in the view config schema versions 1.0.15 and above.
You can diff new and old versions to see the changes.
VitessceConfig constructor
The VitessceConfig constructor now requires schemaVersion as a named parameter.
This will help us to understand which schema version the config was constructed against, enabling better warning messages and error handling.
For instance, the API may show warnings or throw errors when passed invalid constants based on the specified view config schema version.
New coordination types like obsType would be invalid when using an older schema version such as 1.0.7.
We intend to update the constructors in R and Python to enforce this requirement as well.
For new code, we recommend specifying the latest view config schema version:
const vc = new VitessceConfig({ schemaVersion: "1.0.15", name: "My config" });
For a full list of valid schemaVersion values, see the table of schema versions.
Next steps
We hope that these changes make it easier to configure, prepare data, and develop plugins for visualizing one or more related single-cell experiments with Vitessce. While we want our tools to be general enough to facilitate many use cases, we recognize the danger of over abstraction (e.g., preventing users from easily configuring Vitessce), and hope that our implementation strikes a useful balance. We welcome your feedback via issues and discussions.
Some additional use cases that we are excited about:
- using
obsType: 'nucleus'to accurately refer to observations from single-nucleus experiments (similarlyobsType: 'nucleolus',obsType: 'mitochondria', etc. as technologies for isolating subcellular components mature) - using
featureType: 'isoform'to accurately refer to features from isoform-resolved sequencing- and FISH-based experiments - using
featureType: 'topic'andfeatureValueType: 'probability'to enable visualization of topic model-based analysis results
Demos
The following examples showcase how the updates described here can be used to perform integrative visualization of single-cell data.
Migration
The Vitessce JavaScript, Python, R packages will continue to be backwards compatible with previous view config schema versions
(i.e., using version: '1.0.1' in a JSON view config would result in the previously documented behavior despite upgrading the Vitessce package version).
For developers looking to upgrade to the latest version of the vitessce JavaScript package, please visit the upgrade guide.